UDI et sérialisation des dispositifs médicaux
La date limite pour la mise en œuvre de la traçabilité des dispositifs médicaux dans l’Union européenne approche. D’ici à la fin 2027, les dispositifs à haut risque (classe 3) devront être marqués d’une UDI et d’ici à la fin 2028, les dispositifs à faible et moyen risque (classes 1 et 2) devront également être conformes.
Présentation de l’IUD
Le sigle IUD désigne « l’identifiant unique des dispositifs » et fait référence aux dispositifs médicaux. La Food and Drug Administration (FDA) américaine et la Commission européenne ont toutes deux légiféré en la matière. Ces lois concernent la gamme complète des dispositifs médicaux de la classe 1 (risque faible, comme les bandages, etc.) à la classe 3 (dispositifs à haut risque pour appliquer les mesures de maintien des fonctions vitales, par exemple : stimulateurs cardiaques), en passant par la classe 2 (risque modéré, par exemple : appareils auditifs, cathéters). Le règlement relatif à l’IUD exige que le dispositif médical en lui-même ainsi que tous les niveaux de conditionnement supérieurs (pas les unités logistiques) soient identifiés par un code IUD unique, dans un format lisible par machine (code-barres) et par l’humain. Un marquage direct sur le produit lui-même est nécessaire pour les dispositifs médicaux destinés à une utilisation répétée sur une longue période et qui sont retraités plusieurs fois, car ils sont obligatoirement séparés de leur emballage d’origine. Pour appliquer le codage IUD, différentes technologies (jet d’encre thermique, transfert thermique ou laser) sont disponibles en fonction du matériau.
Objectifs du règlement relatif à l’IUD
- Mettre en place un système d’identification adéquat des dispositifs médicaux à travers la chaîne d’approvisionnement et lors de leur utilisation.
- Avoir accès à des informations importantes et complètes sur le dispositif médical.
- Pouvoir documenter de manière standardisée l’utilisation des dispositifs médicaux dans les dossiers médicaux électroniques.
Chaque autorité a créé une base de données à cet effet. Aux États-Unis, il s’agit de la base de données GUDID et au sein de l’Union européenne, de la base de données EUDAMED. Le fabricant est désormais tenu d’enregistrer chaque produit avec ses éléments de données correspondants dans la base de données des autorités.
Structure du code IUD

Les termes et abréviations entourant le règlement relatif à l’IUD peuvent être très déroutants. Voici tout d’abord quelques définitions :
IUD = identifiant unique des dispositifs – codage sur le produit ; correspond à la norme GS1. L’IUD se compose de l’ID et de l’IP.
ID = identifiant des dispositifs
IP = identifiant « production » – autres identifiants relatifs à la production, notamment :
- Lot : AI(10)
- Date de péremption : AI(17)
- Numéro de série : AI(21).
Le fabricant décide des informations qui seront utilisées.
=> Le codage IUD se compose donc du code GTIN et des informations facultatives relatives à la production.
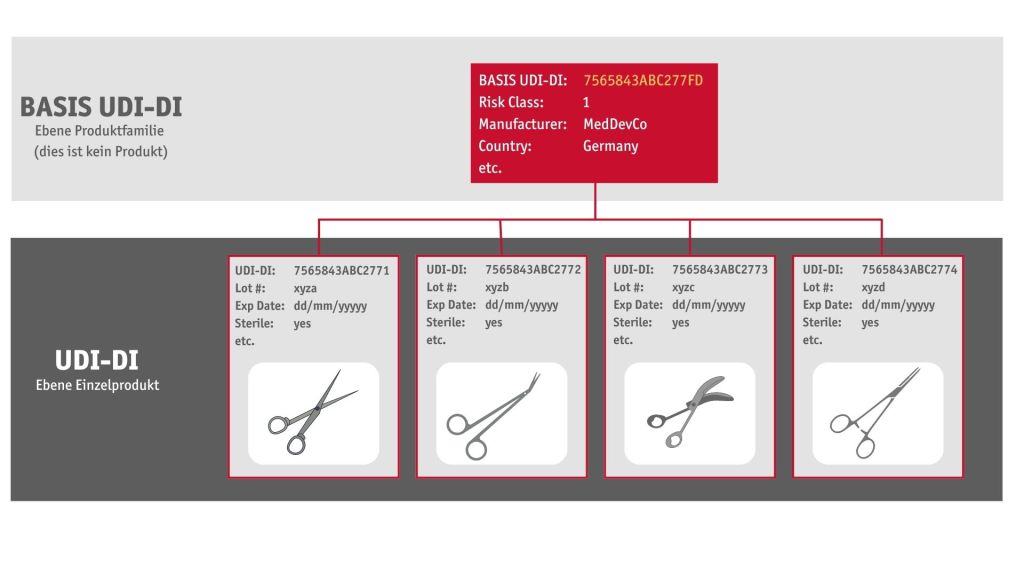
Contrairement aux États-Unis, l’Europe exige « l’IUD-ID de base ». L’IUD-ID de base identifie une gamme de produits complète. Outre cet IUD-ID de base, chaque produit de cette gamme possède son propre numéro unique. Ce numéro est l’IUD-ID. L’IUD-ID de base apparaît dans la base de données EUDAMED et dans les documents officiels. Il n’apparaît jamais sur le produit ou son emballage.
L’IUD-ID (code GTIN) apparaît dans la base de données EUDAMED, et sur les produits ou leur emballage. Sur la base de l’IUD-ID (code GTIN), le produit est ensuite identifié dans la base de données des autorités, et toutes les informations correspondantes sont mises à la disposition du demandeur.
Réglementations : dispositifs médicaux et produits pharmaceutiques
En général, le suivi et la traçabilité sont actuellement uniquement nécessaires pour les produits pharmaceutiques. En mettant en place et en utilisant un système d’identification, les entreprises qui fabriquent des produits pharmaceutiques et des dispositifs médicaux deviennent porteuses des informations nécessaires au suivi et à la traçabilité. Ces informations peuvent être utilisées pour identifier chaque article. Mais certaines initiatives planchent déjà sur une recommandation visant à traiter les dispositifs médicaux de la même manière que les produits pharmaceutiques. Nous pensons que dans un avenir proche, les fabricants de dispositifs médicaux adopteront le modèle pharmaceutique et devront automatiser les informations de suivi des produits.
C’est pourquoi nous recommandons dès aujourd’hui à tous les fabricants de dispositifs médicaux d’utiliser un système de suivi et de traçabilité Laetus, même s’il n’y a pas encore d’exigences directes de sérialisation. La société Laetus dispose d’une expérience de plusieurs dizaines d’années dans le domaine pharmaceutique et, grâce à sa vaste clientèle pharmaceutique, constitue le partenaire idéal pour faire vos premiers pas en matière de suivi et de traçabilité.
Cela dit, même en l’absence de sérialisation, l’utilisation d’un système de suivi et de traçabilité Laetus peut apporter une valeur ajoutée au fabricant de dispositifs médicaux à de nombreux égards. En particulier pour les fabricants qui produisent des dispositifs en série sur de nombreuses lignes de production réparties sur plusieurs sites, il s’avère judicieux d’utiliser un système supérieur qui distribue automatiquement les commandes aux lignes de production, et fournit les données correctes aux imprimantes et caméras correspondantes. Les erreurs dues au chargement d’un format erroné par l’opérateur sont éliminées. Le système peut communiquer directement avec votre ERP pour recevoir, traiter et confirmer les données des commandes. Des comptes-rendus et des enregistrements de modifications uniformes améliorent la qualité de la production.
Pourquoi la sérialisation est-elle utile ?
La sérialisation et l’agrégation permettent de suivre le produit tout au long de la chaîne. Il est important de savoir combien de produits ont été fabriqués et où ils se trouvent dans la chaîne, surtout lorsque le débit est élevé. La traçabilité des produits apporte donc une forte valeur ajoutée interne.
Le système utilisé doit impérativement respecter les réglementations relatives à l’IUD et les réglementations pharmaceutiques, et garantir la plus grande flexibilité possible. Lors de la conception d’un système de soutien à la sérialisation, il est donc judicieux d’inclure les exigences liées à l’IUD et vice versa. Si une future exigence d’automatisation de l’échange d’informations est adoptée, la structure de sérialisation créée pour les médicaments pourra également être utilisée pour les dispositifs médicaux. Nous recommandons une approche globale pour le développement de votre système IUD, car la sécurité de la chaîne d’approvisionnement et les éventuelles contrefaçons représenteront un problème permanent.
Conclusion
En résumé, les organisations devraient développer leur stratégie de mise en œuvre de l’IUD en tenant compte de la situation dans son ensemble et développer des solutions qui répondent aux exigences immédiates, mais qui tiennent également compte des exigences potentielles en matière de suivi et de traçabilité. La mise en place d’une solution flexible peut nécessiter un investissement important à court terme, mais peut permettre de réaliser des économies considérables par la suite.